

Molecular alterations of gastrointestinal stromal tumor - Prognostic and therapeutic implications
Sun, 2017-08-27 10:27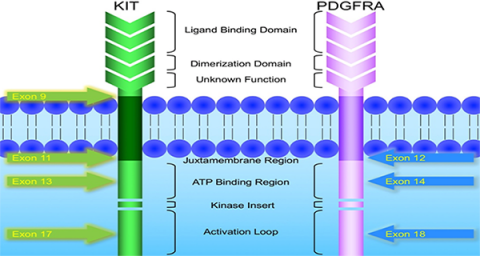
Molecular alterations of gastrointestinal stromal tumor - Prognostic and therapeutic implications
Volume 12, Issue 2 August 2017 (download full article in pdf)
Editorial note:
Gastrointestinal stromal tumor is the commonest mesenchymal tumor in the digestive system. It is a genetically heterogeneous disease with various mutations apart from classical activation mutations in KIT and PDGFRA genes. In the topical update, Dr. Anthony Chan provided an overview of molecular alterations of gastrointestinal stromal tumor with emphasis on their prognostic and therapeutic significance. We welcome any feedback or suggestions. Please direct them to Dr. Anthony Chan (e-mail: awh_chan@cuhk.edu.hk) of Education Committee, the Hong Kong College of Pathologists. Opinions expressed are those of the authors or named individuals, and are not necessarily those of the Hong Kong College of Pathologists.
Dr. Anthony W.H. Chan
Clinical Associate Professor
Department of Anatomical and Cellular Pathology, Prince of Wales Hospital, The Chinese University of Hong Kong
The gist of GIST
Gastrointestinal stromal tumor (GIST) is a rare tumor with the annual incidence rate of 10- 15/1,000,000, but it is the commonest mesenchymal tumor in the digestive system. It affects both sexes equally and presents at any age from children to elderly with the median age of mid 60s. Stomach (55.6%) is the most frequent primary tumor site followed by small intestine (31.8%), large intestine (6.0%) and esophagus (0.7%). Other uncommon primary sites, such as omentum, mesentery and liver, accounts for 5.5% of all GISTs.(1) Important milestones of GIST in diagnostic, prognostic and therapeutic aspects are briefly summarized in this section.
In the past, GIST was regarded as leiomyoma, leiomyoblastoma or leiomyosarcoma before the era of wide availability of immunohistochemistry. In 1983, Mazur and Clark first applied the term "stromal tumor" to describe a group of gastric mesenchymal tumor lacking ultrastructural features of smooth muscle or schwann cells.(2) In 1989, a short-lived term, gastrointestinal autonomic nerve tumor (GANT), was used to describe a small subset of GIST featured by small intestinal location, epithelioid appearance and focal immunoreactivity towards neural markers (S100, neurofilament and synaptophysin).(3) In 1995, CD34 was found to be the first useful diagnostic immunohistochemical marker to differentiate GIST from leiomyoma and schwannoma although only 60-70% of all GISTs are immunoreactive to CD34.(4) In 1998, the hallmark constitutive activation mutation of KIT gene and overexpression of KIT/CD117 protein in GIST were discovered by Hirota et al.(5) This finding also suggested that GIST may be originated from interstitial cells of Cajal, pacemaker cells of intestine, which express KIT and CD34. However, activation mutation of KIT gene and overexpression of KIT are not consistently correlated. A subset of KIT positive GISTs was found to lack KIT mutation and this observation led to the subsequent discovery of gain-of-function mutation of platelet-derived growth factor receptor alpha (PDGFRA) gene in 2003.(6, 7) KIT and PDGFRA mutations are mutually exclusive. About 5-10% of GISTs, particularly those with PDGFRA mutation do not express KIT. In 2004, West et al. identified a novel gene, DOG1 (discovered on GIST-1), through cDNA microarray, and showed DOG1 protein was highly expressed in GISTs (97.8%), including those KIT negative GISTs.(8) KIT and/or DOG1 become crucial diagnostic immunohistochemical markers for GIST. A small subgroup of GISTs with immunoreactivity of KIT/DOG1 lack neither KIT or PDGFRA mutation was first designated as wild-type GISTs in the same year.(9) Wild-type GISTs are later shown to be a heterogeneous group with various mutations.(10- 13)
Prognosis of patients with GIST is shown to be correlated with tumor size and mitosis. The first consensus risk stratification was proposed by investigators in National Institutes of Health (NIH) in 2002 (Table 1).(14) Anatomical location of GIST is also an important prognostic factor and firstly integrated to the Armed Forces Institute of Pathology system in 2006 (Table 2)(15). Gastric GIST behaves more indolent than small and large bowel GIST with similar size and mitosis. Tumor rupture is an additional prognosticator for GIST patients and incorporated into the modified NIH system in 2008 (Table 3).(16) Finally, the most widely adopted tumor staging system, American Joint Committee on Cancer (AJCC), include GIST risk stratification composed of tumor size, mitosis, anatomical location, nodal and distant metastases in the 7th edition in 2010, which remains unchanged in the recently released 8th edition (Table 4 and 5).
Surgical resection remains the mainstay of curative therapy for GIST but a substantial portion of GIST patients present in advanced stage beyond surgical intervention. Imatinib, a multi-targeted tyrosine kinase inhibitor specific for c-abl, c-kit and PDGFR, was first used in a patient with metastatic GIST in 2001.(17) The dramatic clinical response from this patient and the subsequent successful phase II clinical trial in 2002 secured the first-line role of imatinib for patients with inoperable GIST and pioneered molecular targeted therapy for sarcoma.(18) Primary and acquired resistance to imatinib among GIST patients led to development of newer targeted agents. Two hallmark phase III randomized controlled trials on sunitinib (NCT00075218) and regorafenib (NCT01271712) for GIST were completed in 2006 and 2013, respectively.(19, 20) Sunitinib and regorafenib are indicated for patients with advanced GIST resistant or intolerant to imatinib.
Table 1: NIH risk stratification for GIST (14)
|
GROUP |
SIZE (CM) |
MITOSIS (/50 HPF) |
|---|---|---|
| Very low risk | <2 | ≤5 |
| Low risk | 2-5 | ≤5 |
| Intermediate risk | <5 | 6-10 |
| 5-10 | ≤5 | |
| High risk | >5 | >5 |
| >10 | Any | |
| Any | >10 |
Table 2: AFIP risk stratification for GIST (15)
| GROUP | SIZE (CM) | MITOSIS (/50 HPF) | STOMACH | DUODENUM | JEJUNUM /ILEUM | RECTUM |
|---|---|---|---|---|---|---|
| 1 | ≤2 | ≤5 | None | None | None | None |
| 2 | >2-5 | ≤5 | Very low | Low | Low | Low |
| 3a | >5-10 | ≤5 | Low | Moderate | - | - |
| 3b | >10 | ≤5 | Moderate | High | High | High |
| 4 | ≤2 | >5 | None | High | - | High |
| 5 | >2-5 | >5 | Moderate | High | High | High |
| 6a | >5-10 | >5 | High | High | - | - |
| 6b | >10 | >5 | High | High | High | High |
Table 3: Modified NIH risk stratification for GIST (16)
| GROUP | SIZE (CM) | MITOSIS (/50 HPF) | PRIMARY SITE |
|---|---|---|---|
| Very low risk | ≤2 | ≤5 | Any |
| Low risk | >2-5 | ≤5 | Any |
| Intermediate Risk | >2-5 | >5 | Gastric |
| ≤5 | 6-10 | Any | |
| >5-10 | ≤5 | Gastric | |
| High risk | >5 | >5 | Any |
| >10 | Any | Any | |
| Any | >10 | Any | |
| Any | Any | Tumor rupture | |
| >2-5 |
>5
|
Non-gastric | |
| >5-10 | ≤5 | Non-gastric |
Table 4: AJCC staging system for gastric and omental GIST
| GROUP | SIZE (CM) | N | M | MITOSIS |
|---|---|---|---|---|
| IA | ≤5 | 0 | 0 | ≤5 |
| IB | >5-10 | 0 | 0 | ≤5 |
| II | ≤5 | 0 | 0 | >5 |
| >10 | 0 | 0 | ≤5 | |
| IIIA | >5-10 | 0 | 0 | >5 |
| IIIB | >10 | 0 | 0 | >5 |
| IV | Any | 1 | 0 | Any |
| Any | Any | 1 | Any |
Table 5: AJCC staging system for small/large bowel, esophageal, mesenteric and peritoneal GIST
| GROUP | SIZE (CM) | N | M | MITOSIS |
|---|---|---|---|---|
| I | ≤5 | 0 | 0 | ≤5 |
| II | >5-10 | 0 | 0 | ≤5 |
| IIIA | ≤2 | 0 | 0 | >5 |
| >10 | 0 | 0 | ≤5 | |
| IIIB | >2 | 0 | 0 | >5 |
| IV | Any | 1 | 0 | Any |
| Any | Any | 1 | Any |
Mutational landscape of GIST
KIT and PDGFRA mutations are major driver mutations in GIST tumorigenesis. Both genes encode type III receptor tyrosine kinases with similar structures: extracellular ligand binding domain and dimerization domain, a transmembrane sequence, a juxtamembrane domain and intracellular kinase domain (Figure 1). Binding of corresponding ligands, stem cell factor and PDGFA, to c-kit and PDGFRA receptor, respectively, dimerizes and activates receptor tyrosine kinases. In GIST, activation mutations in KIT and PDGFRA lead to uncontrolled ligand- independent receptor activation. Mutation hotspots of KIT gene are located at exons 9, 11, 13 and 17, whereas those of PDGFRA gene are situated at exons 12, 14 and 18. Mutation of extracellular domain of KIT encoded by exon 9 facilitate receptor dimerization. Mutations in the juxtamembrane domain, which is encoded by exon 11 of KIT and exon 12 of PDGFRA, allow dimerization of receptor without binding of ligands. Mutations of ATP binding region of kinase domain (encoded by exon 13 of KIT and exon 14 of PDGFRA) enhance kinase activity, while mutations of activation loop (encoded by exon 17 of KIT and exon 18 of PDGFRA) promote active conformation of kinase.(21) Table 6 and Figure 2 summarize the mutational landscape of GIST based on the data from population-based studies and clinical trials.(22-29) Frequencies of PDGFRA mutations are significantly lower among patients in clinical trials (mean 1.7%) than those in population-based studies (mean 14.9%) because GIST patients with PDGFRA mutations are associated with better prognosis and earlier stage and hence do not require systemic therapy.(9, 22, 23, 29)
KIT mutation accounts for 71.5% (64.8-89.1%) of mutations in GISTs.(24, 25, 27-29) Exon 11 mutation is the commonest mutation (61.1%, range: 56.1-77.1%). Deletion, substitution and duplication contribute to 23-28%, 2-20% and 2-7%, respectively. Deletion in exon 11 is associated with younger age, larger tumor size, higher mitotic count and poor prognosis, whereas duplication is associated with female and stomach predilection and better prognosis. Exon 9 mutation is found in 7.1-10.9% of GISTs, particularly in those arising from small and large intestine, and associated with poor prognosis. Exon 13 and exon 17 are rare mutation hotspots (<1-2%) in GISTs, which are almost exclusively spindle in morphology and more frequently developed in small intestine. GISTs with exon 13 and 17 mutants are associated good and intermediate prognosis, respectively.
PDGFRA mutation accounts for 14.9% (4.7-21.1%) of mutations in GISTs.(24, 25, 27-29) About 30- 40% of GISTs without immunoreactivity of KIT/CD117 harbour PDGFRA mutation. GISTs with PDGFRA mutation generally show predilection to gastric location (>90%) and epithelioid/mixed morphology, and favourable prognosis (except non-D842V exon 18 mutation).
Wild-type GIST, which express immunoreactivity of KIT/DOG1 but lack neither KIT or PDGFRA mutation, contributes to 13-18% of adult GISTs and 85% of pediatric GIST.(10-12) As previously mentioned, it is a genetically heterogeneous group (Figure 3). Wild-type GIST can be further stratified by using succinate dehydrogenase B (SDHB) immunohistochemistry and familial syndromes. On one hand, SDHB deficient wild-type GISTs accounts for about 5% of all GISTs, and can be sporadic or related to Carney triad and Carney- Stratakis syndrome. Carney triad is a constellation of GIST, paraganglioma and pulmonary chondroma with undetermined germline mutation, whereas Carney-Stratakis syndrome is an autosomal dominant disease with dyad of GIST and paraganglioma, and germline mutations in SDHB, SDHC or SDHD genes.(30) SDHB deficient wild-type GISTs are featured by female predominance (except for Carney-Stratakis syndrome), exclusive location in stomach, multifocality, epithelioid/mixed morphology, unpredictable clinical outcome by histology, indolent clinical course despite frequent nodal metastasis, and mutation id SDH subunits (except for Carney triad). On the other hand, SDHB proficient wild-type GISTs make up 10.5% of all GISTs, and are either sporadic (9%) or syndromic (1.5%). Syndromic SDHB proficient wild-type GISTs are associated with neurofibromatosis type 1, absence of sex/age predilection, small intestine in location, multifocality, spindle morphology, and favorable prognosis. Sporadic SDHB proficient wild-type GISTs can be further classified according to BRAF mutation. Sporadic SDHB proficient wild-type GISTs with BRAF mutation usually occur in 6th decade of age and small intestine with spindle morphology. Prognosis of this subgroup is inconclusive.(10, 29, 31, 32) Sporadic SDHB proficient wild-type GISTs without BRAF mutation are also known as quadruple wild-type GISTs without any mutation in KIT, PDGFRA, SDH and genes in RAS pathway (BRAF/NF1).(12, 13) They represent the commonest subgroup (7%) of wild-type GISTs and a genetically heterogeneous subgroup harboring ETV6-NTRK3 translocation, FGFR1-TACC1 translocation, mutation of MEN1 and MAX, and overexpression of COL22A1 and CALCRL.(12, 29, 33, 34) Due to complex genetic heterogeneity, clinicopathological features of this subgroup have not been well characterized.
Figure 1: Schematic diagram of the structures of KIT and PDGFRA receptor tyrosine kinases
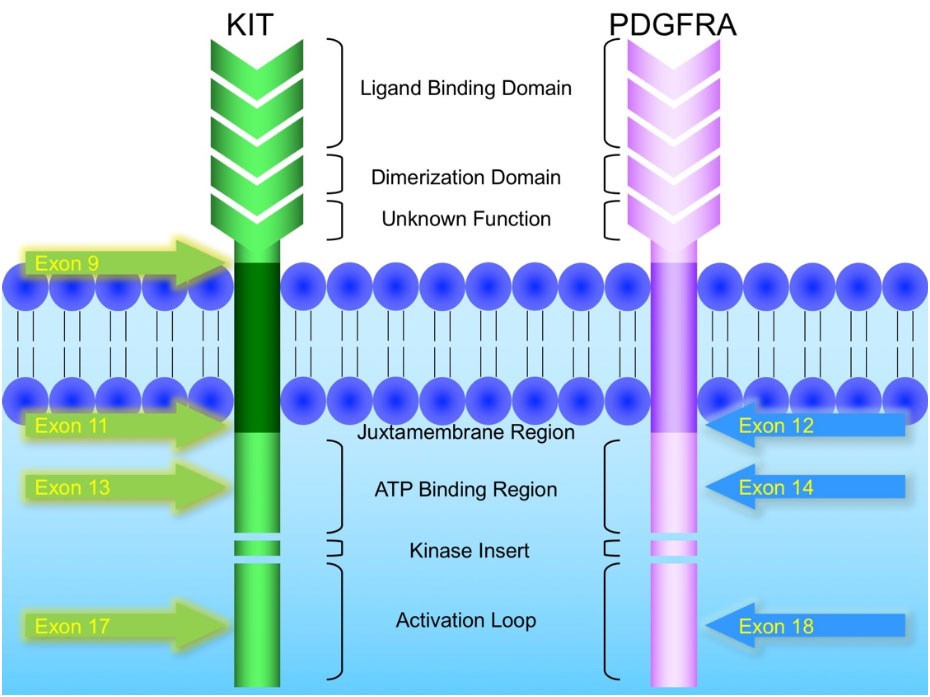
Figure 2: Mutational landscape of GIST

Figure 3: Classification of wild-type GIST
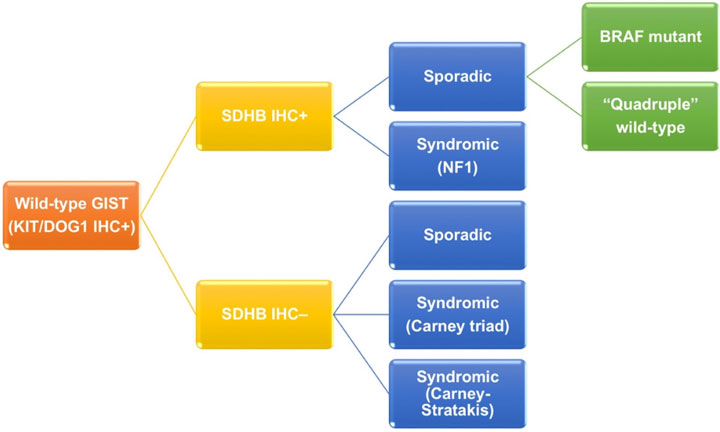
Table 6: Mutational landscape of GIST
| Study | Region | n | KIT exon | PDGFRA exon | Wild type | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 9 | 11 | 13 | 17 | 12 | 14h | 18 | ||||
| Wozniak 2012 (24) | Poland | 427 | 7.3% | 61.1% | 0.5% | 0.5% | 0.2% | 0.7% | 11.9% | 17.8% |
| Wozniak 2014 (28) | Europe | 1056 | 7.4% | 61.4% | 1.8% | 0.6% | 0.9% | 0.3% | 12.8% | 14.9% |
| Künstlinger 2013 (25) | Germany | 1366 | 9.2% | 59.3% | 1.8% | 0.8% | 1.8% | 0.6% | 13.8% | 12.7% |
| Wang 2014 (27) | China | 275 | 10.9% | 77.1% | 1.1% | 0.0% | 1.1% | 0.0% | 3.6% | 6.2% |
| Rossi 2015 (29) | Italy | 451 | 7.1% | 56.1% | 0.9% | 0.7% | 2.2% | 1.6% | 17.3% | 14.2% |
| ACOSOG Z9001 (26) | 507 | 6.9% | 67.3% | 1.8% | 0.2% | NA | NA | NA | 12.8% | |
| CALGB 150105 (23) | 378 | 8.2% | 72.8% | 0.8% | 1.1% | 0.0% | 0.0% | 1.6% | 15.3% | |
| EORTC 62005 (22) | 377 | 15.4% | 65.8% | 1.6% | 0.8% | 0.8% | 0.0% | 1.9% | 13.8% | |
Clinical implications of mutations in GIST
Different mutations in GIST have their own characteristic prognostic and therapeutic implications. Prognostic significance of individual mutations have been described by various investigators and briefly mentioned in the previous section. Rossi et al. recently systemically analyzed the prognostic impact of mutations among 451 patients with primary localized treatment-naive GISTs.(29) By multivariable Cox regression, mutational status was an independent prognosticator in addition to patient's age, tumor location, tumor size and mitotic count. Three molecular risk groups with prognostic significance were identified: Group 1 with the most favorable outcome is composed of mutations in KIT exon 13, PDGFRA exon 12 and BRAF; Group 2 with the intermediate outcome (hazard ratio 3.06) consists of KIT/PDGFRA/BRAF triple negative, and mutations in KIT exon 17, PDGFA exon 14 and 18 (D842V); and Group 3 with the most unfavorable outcome comprises mutations in KIT exon 9 and 11, and PDGFRA exon 18 (non-D842V).
Clinical response toward imatinib among GIST patients is closely related to tumor genotype. In a phase III clinical trial (SWOG S0033/CALGB 150105), the investigators demonstrated that patients with KIT exon 11 mutation (complete response [CR]/partial response [PR] 71.7%) had better response to imatinib than those with KIT exon 9 mutation (CR/PR 44.4%) and wild-type KIT (CR/PR 44.6%).(23) They also showed that doubling the dose of imatinib (from 400 mg to 800 mg) improved response rates for patients with exon 9-mutant tumors (CR/PR 17% vs. 67%). Double dose of imatinib did not offer any better response rate among patients with exon 11 mutant or wild- type KIT. A subsequent meta-analysis of 1,640 patients with advanced GIST receiving imatinib confirmed that double dose of imatinib improved progression-free survival and objective response rate, but not overall survival, among patients with KIT exon 9-mutant GIST.(35) PDGFA exon 18 (D842V) mutation and KIT/PDGFRA wild-type are responsible for primary resistance to imatinib.(36) Among patients with advanced GIST receiving imatinib, a substantial proportion of initial responders will develop acquired resistance. Secondary mutations in exon 11 (L576P and V559A), exon 13 (V654A), exon 14 (T670I), exon 17 and exon 18 (A829P) of KIT, and exon 18 of PDGFRA are related to acquired resistance to imatinib.(36)
Clinical response to sunitinib, the second line targeted therapy after imatinib failure, is also considerably affected by primary and acquired mutations of KIT. Patients with primary KIT exon 9 mutation or wild-type KIT had better overall and progression-free survival than those with KIT exon 11 mutation, whereas patients with acquired KIT exons 13 or 14 mutations had better outcome than those with KIT exon 17 or 18 mutations.(37) Similarly, clinical response to regorafenib, the third line therapy after imatinib and sunitinib failure, is significantly influenced by tumor genotype. Regorafenib provided better clinical outcome among patients with primary KIT exon 11 mutation and SDHB deficient GIST, (38) as well as those with secondary mutation of KIT exon 17, which are resistant to both imatinib and sunitinib.(39)
Summary
GIST is a genetically heterogeneous tumor. Genotypes and phenotypes are closely interrelated. Specific mutations have their characteristic clinicopathological features, prognostication and therapeutic implications. Genetic analyses KIT and PDGFRA are highly recommended especially among patients with advanced diseases undergoing targeted therapy. Wild-type GISTs are recommended to be further analysed by SDHB immunohistochemistry and BRAF mutation test.
Reference
1. Soreide K, Sandvik OM, Soreide JA, Giljaca V, Jureckova A, Bulusu VR. Global epidemiology of gastrointestinal stromal tumours (GIST): A systematic review of population-based cohort studies. Cancer Epidemiol. 2016;40:39-46.
2. Mazur MT, Clark HB. Gastric stromal tumors. Reappraisal of histogenesis. Am J Surg Pathol. 1983;7(6):507-19.
3. Herrera GA, Cerezo L, Jones JE, Sack J, Grizzle WE, Pollack WJ, et al. Gastrointestinal autonomic nerve tumors. 'Plexosarcomas'. Arch Pathol Lab Med. 1989;113(8):846-53.
4. Miettinen M, Virolainen M, Maarit Sarlomo R. Gastrointestinal stromal tumors--value of CD34 antigen in their identification and separation from true leiomyomas and schwannomas. Am J Surg Pathol. 1995;19(2):207- 16.
5. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain- of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577-80.
6. Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708-10.
7. Hirota S, Ohashi A, Nishida T, Isozaki K, Kinoshita K, Shinomura Y, et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology. 2003;125(3):660-7.
8. West RB, Corless CL, Chen X, Rubin BP, Subramanian S, Montgomery K, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol. 2004;165(1):107-13.
9. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22(18):3813-25.
10. Agaram NP, Wong GC, Guo T, Maki RG, Singer S, Dematteo RP, et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2008;47(10):853-9.
11. Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011;108(1):314-8.
12. Nannini M, Astolfi A, Urbini M, Indio V, Santini D, Heinrich MC, et al. Integrated genomic study of quadruple-WT GIST (KIT/PDGFRA/SDH/RAS pathway wild-type GIST). BMC Cancer. 2014;14:685.
13. Pantaleo MA, Urbini M, Indio V, Ravegnini G, Nannini M, De Luca M, et al. Genome-Wide Analysis Identifies MEN1 and MAX Mutations and a Neuroendocrine-Like Molecular Heterogeneity in Quadruple WT GIST. Mol Cancer Res. 2017;15(5):553-62.
14. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33(5):459-65.
15. Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol. 2006;23(2):70-83.
16. Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol. 2008;39(10):1411-9.
17. Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344(14):1052-6.
18. Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472-80.
19. Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-38.
20. Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302.
21. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865-78.
22. Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42(8):1093- 103.
23. Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26(33):5360-7.
24. Wozniak A, Rutkowski P, Piskorz A, Ciwoniuk M, Osuch C, Bylina E, et al. Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): Polish Clinical GIST Registry experience. Ann Oncol. 2012;23(2):353-60.
25. Kunstlinger H, Huss S, Merkelbach-Bruse S, Binot E, Kleine MA, Loeser H, et al. Gastrointestinal stromal tumors with KIT exon 9 mutations: Update on genotype-phenotype correlation and validation of a high-resolution melting assay for mutational testing. Am J Surg Pathol. 2013;37(11):1648-59.
26. Corless CL, Ballman KV, Antonescu CR, Kolesnikova V, Maki RG, Pisters PW, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32(15):1563-70.
27. Wang M, Xu J, Zhao W, Tu L, Qiu W, Wang C, et al. Prognostic value of mutational characteristics in gastrointestinal stromal tumors: a single-center experience in 275 cases. Med Oncol. 2014;31(1):819.
28. Wozniak A, Rutkowski P, Schoffski P, Ray-Coquard I, Hostein I, Schildhaus HU, et al. Tumor genotype is an independent prognostic factor in primary gastrointestinal stromal tumors of gastric origin: a european multicenter analysis based on ConticaGIST. Clin Cancer Res. 2014;20(23):6105-16.
29. Rossi S, Gasparotto D, Miceli R, Toffolatti L, Gallina G, Scaramel E, et al. KIT, PDGFRA, and BRAF mutational spectrum impacts on the natural history of imatinib-naive localized GIST: a population-based study. Am J Surg Pathol. 2015;39(7):922-30.
30. Stratakis CA, Carney JA. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): molecular genetics and clinical implications. J Intern Med. 2009;266(1):43-52.
31. Hostein I, Faur N, Primois C, Boury F, Denard J, Emile JF, et al. BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol. 2010;133(1):141-8.
32. Huss S, Pasternack H, Ihle MA, Merkelbach-Bruse S, Heitkotter B, Hartmann W, et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum Pathol. 2017;62:206-14.
33. Brenca M, Rossi S, Polano M, Gasparotto D, Zanatta L, Racanelli D, et al. Transcriptome sequencing identifies ETV6-NTRK3 as a gene fusion involved in GIST. J Pathol. 2016;238(4):543-9.
34. Shi E, Chmielecki J, Tang CM, Wang K, Heinrich MC, Kang G, et al. FGFR1 and NTRK3 actionable alterations in "Wild-Type" gastrointestinal stromal tumors. J Transl Med. 2016;14(1):339.
35. Gastrointestinal Stromal Tumor Meta- Analysis G. Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28(7):1247-53.
36. Sankhala KK. Clinical development landscape in GIST: from novel agents that target accessory pathways to revisiting non-targeted therapies. Expert Opin Investig Drugs. 2017;26(4):427-43.
37. Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352-9.
38. Ben-Ami E, Barysauskas CM, von Mehren M, Heinrich MC, Corless CL, Butrynski JE, et al. Long-term follow-up results of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosine kinase inhibitor therapy. Ann Oncol. 2016;27(9):1794-9.
39. Yeh CN, Chen MH, Chen YY, Yang CY, Yen CC, Tzen CY, et al. A phase II trial of regorafenib in patients with metastatic and/or a unresectable gastrointestinal stromal tumor harboring secondary mutations of exon 17. Oncotarget. 2017;8(27):44121-30.