

An overview of NMO Spectrum Disorder and the diagnostic utility of anti-NMO antibodies
Fri, 2017-12-22 00:19An overview of NMO Spectrum Disorder and the diagnostic utility of anti-NMO antibodies
Volume 13, Issue 1 December 2017 (download full article in pdf)
Editorial note
NMOSD is an immune mediated demyelinating disease. Though its clinical presentation may overlap with multiple sclerosis, distinguishing these two entities is important in view of differences in treatment. Anti-NMO antibodies play a crucial role in the workup and diagnosis of NMOSD. In this review, Dr Elaine Au provided an overview of the NMOSD condition and the use of anti-NMO antibody assays. We welcome any feedback or suggestions. Please direct them to Dr Elaine Au (email: ayl436@ha.org.hk) of Education Committee, the Hong Kong College of Pathologists. Opinions expressed are those of the authors or named individuals, and are not necessarily those of the Hong Kong College of Pathologists.
Dr Au Yuen Ling Elaine
Associate Consultant, Division of Clinical Immunology, Department of Pathology, Queen Mary Hospital
NMO is an idiopathic immune mediated demyelinating disease that predominantly affects optic nerves and spinal cord. The prevalence range from 0.3 to 4.4 per 100000 people, with Asian and African-American more affected than Caucasian, where multiple sclerosis is more common in the white population (1-5). The condition has been named as Devic’s disease in the past (6), which described a monophasic disorder presenting with simultaneous bilateral optic neuritis and transverse myelitis. With the availability of specific serological marker, antibodies that targeted the water channel aquaporin-4 (AQP4), the clinical and neuroimaging spectrum of NMO disease is broadened. Instead of being a monophasic disorder, NMO antibodies positive patients with recurrent attacks are not uncommonly found. Moreover, the clinical presentations are more variable than the traditional Devic disease. There is no pathognomonic clinical feature of spectrum disorder (NMOSD), though certain clinical presentations are particularly suggestive of the disorder. These include simultaneously bilateral optic neuritis, complete spinal cord syndrome and area postrema clinical syndrome.
Multiple sclerosis is an important differential diagnosis of this condition in view of the overlapping clinical features of these two conditions. In NMOSD, optic neuritis tends to be more severe, more often with simultaneous bilateral involvement or sequential in rapid succession, compare to multiple sclerosis. Complete spinal cord syndrome, with longitudinally extensive transverse myelitis in MRI, is more suggestive of NMOSD than multiple sclerosis. Differentiating NMOSD from other demyelinating disease, i.e. multiple sclerosis is important since the treatment is different. Indeed, some multiple sclerosis therapies may aggravate NMO disorders (7-10).
Diagnostic Criteria
In 2006, the serological marker was first incorporated into the revised NMO diagnostic criteria. In 2007, NMOSD was introduced to include seropositive patients who do not follow the classical monophasic bilateral optic neuritis and transverse myelitis. Lately, the International Panel for NMO Diagnosis (IPND) has further revised the diagnostic criteria (11). For anti-NMO antibodies positive cases, presenting at least one core clinical characteristic of the disease is required for diagnosis. Core clinical characteristics include optic neuritis, acute myelitis, area postrema syndrome (episode of otherwise unexplained hiccups or nausea and vomiting), acute brainstem syndrome, narcolepsy or acute diencephalic syndrome with typical Magnetic Resonance Imaging (MRI) lesions and symptomatic cerebral syndrome with typical MRI lesions. On the other hand, more stringent clinical criteria, with additional neuroimaging findings, are required in seronegative patients to fulfill the diagnostic criteria (See appendix).
For the workup of the disease, MRI, cerebral spinal fluid (CSF) exam and serological test for anti-NMO antibodies are important. In some patients, CSF pleocytosis, usually in the form of monocytosis and lymphocytosis, are present. Increased CSF protein levels are noted in 46-75% of cases (12, 13). Nevertheless, presence of CSF oligoclonal bands is uncommon in NMOSD, and at most transient, in contrast to the presence of persistent CSF oligoclonal bands in the case of multiple sclerosis. Finally, visual evoked potentials, somatosensory evoked potentials and brainstem acoustic evoked potentials examination may also be helpful in the workup.
Clinical course and prognosis
In NMOSD syndromes affecting regions other than optic nerve and spinal cord, not uncommonly patients will relapse with more classical involvement in subsequent attacks. Majority of NMOSD patients suffer from recurrent attacks (80- 90%), less frequently monophasic attack (10-20%) (14), while gradual progressive course with neurological deterioration is very rare (15). Relapses usually occur in clusters, but unpredictable intervals. In the Mayo Clinic series, the second relapse occurred within 1 year in 60% of cases, and within 3 years in 90% of cases (16). Repeated NMO attacks not uncommonly lead to accumulation of neurological impairment. NMO is associated with more adverse outcome than MS in general(14).
NMOSD has been shown to be frequently associated with other autoimmune disorders, including lupus, Sjogren’s syndrome, etc (17-19). On contrary, NMOSD is not common in rheumatic disease patients.
Anti-NMO antibodies
Among the different investigations, NMO antibodies test is central to the workup. Anti-NMO antibodies are pathogenic. The third extracellular loop of AQP4 is the major epitope for the anti- NMO antibodies. Biopsy and autopsy tissue obtained from seropositive patients demonstrate loss of AQP4 immunoreactivity. Perivascular complement activation in actively demyelinating lesions is also happened. In the central nervous system (CNS), AQP4 is expressed at the foot processes of astrocytes, near the basement membranes, in the optic nerve, in a subpopulation of ependymal cells, in hypothalamic nuclei and in the subfornical organ (20, 21). Truncated astrocyte processes or cell loss were found in demyelinating lesions (11). In rat models, passive transfer of the antibodies leaded to the development of disease (22, 23). These pathological findings distinguish NMOSD from multiple sclerosis.
The Anti-NMO antibodies can be detected by indirect immunofluorescence staining on tissue slide using mouse cerebellum tissue section, cell- based assays, radioimmunoprecipitation assays and enzyme-linked immunosorbent assays (ELISA). Overall, cell-based assay is preferred in view of better assay sensitivity and specificity compared to other methods. Ideally, confirmatory testing with one or more techniques is suggested (11), especially in cases with atypical presentation or borderline results are obtained.
In tissue based indirect immunofluorescence test, NMO antibodies positive case is characterized by the binding to structures adjacent to microvasculature, the Virchow-Robin spaces (VRS) and pia mater (Fig.1).
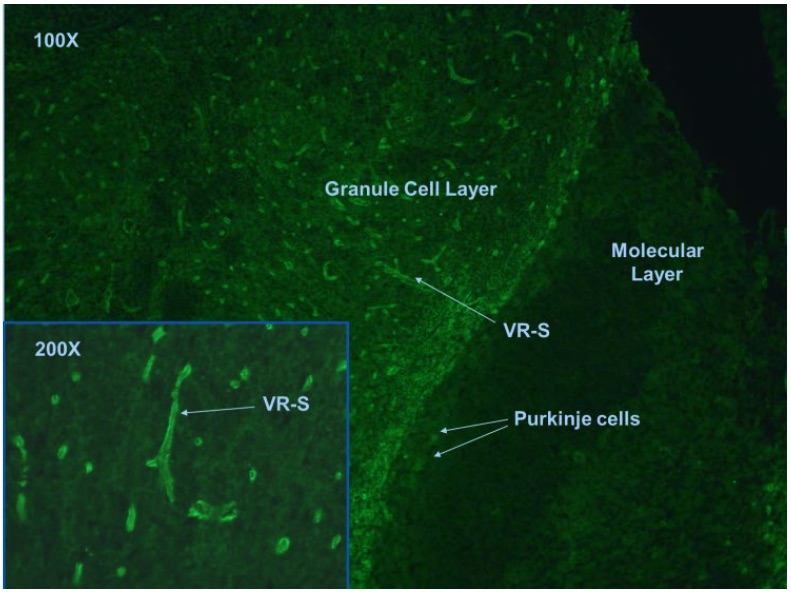
Fig. 1. Mouse cerebellum tissue section stained with anti-NMO antibodies.
This assay allows the detection of any coexisting anti-neuronal antibodies, which may be important as differential diagnosis and workup. However, the method is observer dependent and subjective. The interpretation of antibody staining may easily be affected by non-specific background staining on the tissue. In some rare occasions, antibodies other than anti-NMO may mimic the staining pattern and lead to false positive results. Moreover, indirect tissue based immunofluorescence test has relative low sensitivity (63-64%) (24-27).
Cell-based assay utilize cell lines such as human embryonic kidney 293 (HEK293) cells or Chinese hamster ovary (CHO) cells that have been transfected with AQP4 gene expression vector, so that expressing much higher level of antigen comparing to normal tissues. Cell lines from different units may use different ratio of the two isoform of AQP4: M1 and M23 in order to obtain optimal antigen presentation. Cell-based assay can be assessed by indirect immunofluorescence staining or flow cytometry. For indirect immunofluorescence cell- based assay, slide with fixed AQP-4 gene transfected cells and non- transfected cells growing on different biochips are placed side by side for comparison. Therefore, false positivity is minimized with the inclusion of control non-transfected cells. A higher expression of antigen in the transfected cells also enhance the assay sensitivity compared to tissue-based indirect immunofluorescence testing. (Fig.2)
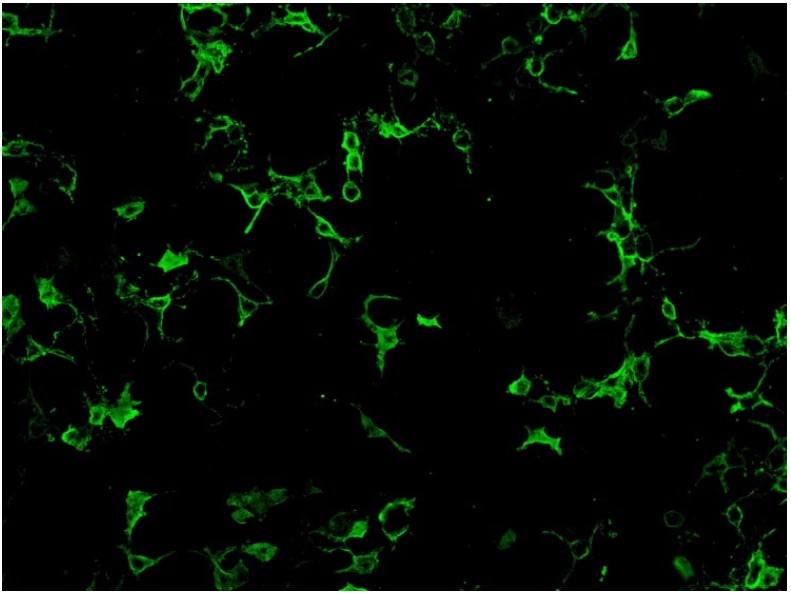
Fig.2. HEK 293 cells transfected with AQP-4 gene expression vector and stained positive with anti- NMO antibodies.
Overall, cell-based assay is the recommended assay in view of good sensitivity and specificity (mean sensitivity 76.7% in a pooled analysis; 0.1% false positivity in a multiple sclerosis cohort) (24- 27). Commercial kits for indirect immunofluorescence cell-based assay are available, which facilitate the assay setup in service laboratories. Nevertheless, indirect immunofluorescence method is semi-quantitative and observer dependent.
Protein-based assays, like ELISA and radioimmunoprecipitation assays, in general, have lower sensitivity compared to cell based assays. In addition, ELISA, in particular at low titer, may yield nonspecific results. However, these assays provide quantitative results which may potentially be used for serial monitoring (24).
Though NMO antibody testing in serum is well- established, the diagnostic role of testing the antibody in CSF is controversial. Most of the cases reported in literature are diagnosed by serum test, though there have been rare cases reported that were CSF positive but serum test negative (28, 29). When studying paired CSF and serum samples with antibody indices calculated, intrathecal production of the NMO antibody is rare (24). NMO antibodies can be present in patients few years before and after the disease presentations. Lately, there is increasing evidence that the antibody titre may reflect disease activity. Elevated antibody levels at relapse and decrease in titre after immunosuppressant treatment has been reported in literature (30-34). Therefore, serial monitoring may possibly facilitate management and medication adjustment. However, there is no general threshold value for clinical relapse and the absolute level varies with individual patients. Rising level may not predict relapse in all cases. In addition, some methodologies, like indirect immunofluorescence test, only provide semi-quantitative results, and inter-run reproducibility is another issue. Other factors including the frequency of test necessary to achieve meaningful disease status monitoring and the cost involved are also important consideration. Therefore, it remains to be determined whether the marker should be serially monitored for treatment response and disease activity monitoring.
Treatment
The treatment for classical Devic’s disease presentation and relapsing NMOSD presentation is no different. High dose steroid is commonly employed as first- line of treatment in acute presentation. Plasma exchange may be considered in treatment refractory cases. Immunomodulatory treatment with interferon β, which is a treatment option in multiple sclerosis, may exacerbate NMOSD disorders. Therefore, differentiating between these two conditions is important. Options of steroid sparing immunosuppressants to consider in NMOSD include azathioprine, methotrexate, mycophenolate mofetil, rituximab, etc (14).
Conclusion
NMOSD is a rare but increasingly recognized condition, which present as an inflammatory and demyelinating autoimmune disorder affecting the central nervous system. With the availability of a serological marker, anti-NMO antibody, the diagnosis and differentiating from related conditions is facilitated. Timely diagnosis and treatment is important for the management of these patients.
Reference
-
Kira J. Multiple sclerosis in the Japanese population. The Lancet Neurology. 2003;2(2):117- 27.
-
abre P, Signate A, Olindo S, Merle H, Caparros-Lefebvre D, Bera O, et al. Role of return migration in the emergence of multiple sclerosis in the French West Indies. Brain : a journal of neurology. 2005;128(Pt 12):2899-910.
-
Asgari N, Lillevang ST, Skejoe HP, Falah M, Stenager E, Kyvik KO. A population-based study of neuromyelitis optica in Caucasians. Neurology. 2011;76(18):1589-95.
-
Cossburn M, Tackley G, Baker K, Ingram G, Burtonwood M, Malik G, et al. The prevalence of neuromyelitis optica in South East Wales. European journal of neurology. 2012;19(4):655-9.
-
Mealy MA, Wingerchuk DM, Greenberg BM, Levy M. Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Archives of neurology. 2012;69(9):1176-80.
-
Jarius S, Wildemann B. The history of neuromyelitis optica. Journal of neuroinflammation. 2013;10:8.
-
Kimbrough DJ, Fujihara K, Jacob A, Lana- Peixoto MA, Leite MI, Levy M, et al. Treatment of Neuromyelitis Optica: Review and Recommendations. Multiple sclerosis and related disorders. 2012;1(4):180-7.
-
Kleiter I, Hellwig K, Berthele A, Kumpfel T, Linker RA, Hartung HP, et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Archives of neurology. 2012;69(2):239-45.
-
Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Multiple sclerosis. 2012;18(1):113-5.
-
Papeix C, Vidal JS, de Seze J,Pierrot- Deseilligny C, Tourbah A, Stankoff B, et al. Immunosuppressive therapy is more effective than interferon in neuromyelitis optica. Multiple sclerosis. 2007;13(2):256-9.
-
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-89.
-
O'Riordan JI, Gallagher HL, Thompson AJ, Howard RS, Kingsley DP, Thompson EJ, et al. Clinical, CSF, and MRI findings in Devic's neuromyelitis optica. Journal of neurology, neurosurgery, and psychiatry. 1996;60(4):382-7.
-
de Seze J, Stojkovic T, Ferriby D, Gauvrit JY, Montagne C, Mounier-Vehier F, et al. Devic's neuromyelitis optica: clinical, laboratory, MRI and outcome profile. Journal of the neurological sciences. 2002;197(1-2):57-61.
-
Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. European journal of neurology. 2010;17(8):1019-32.
-
Wingerchuk DM, Pittock SJ, Lucchinetti CF, Lennon VA, Weinshenker BG. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology. 2007;68(8):603-5.
-
Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology. 1999;53(5):1107-14.
-
Jarius S, Jacobi C, de Seze J, Zephir H, Paul F, Franciotta D, et al. Frequency and syndrome specificity of antibodies to aquaporin-4 in neurological patients with rheumatic disorders. Multiple sclerosis. 2011;17(9):1067-73.
-
Pittock SJ, Lennon VA, de Seze J, Vermersch P, Homburger HA, Wingerchuk DM, et al. Neuromyelitis optica and non organ-specific autoimmunity. Archives of neurology. 2008;65(1):78-83.
-
Wandinger KP, Stangel M, Witte T, Venables P, Charles P, Jarius S, et al. Autoantibodies against aquaporin-4 in patients with neuropsychiatric systemic lupus erythematosus and primary Sjogren's syndrome. Arthritis and rheumatism. 2010;62(4):1198-200.
-
Graber DJ, Levy M, Kerr D, Wade WF. Neuromyelitis optica pathogenesis and aquaporin 4. Journal of neuroinflammation. 2008;5:22.
-
Tait MJ, Saadoun S, Bell BA, Papadopoulos MC. Water movements in the brain: role of aquaporins. Trends in neurosciences. 2008;31(1):37-43.
-
Kinoshita M, Nakatsuji Y, Moriya M, Okuno T, Kumanogoh A, Nakano M, et al. Astrocytic necrosis is induced by anti-aquaporin-4 antibody- positive serum. Neuroreport. 2009;20(5):508-12.
-
Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T, et al. Neuromyelitis optica: Passive transfer to rats by human immunoglobulin. Biochemical and biophysical research communications. 2009;386(4):623-7.
-
Jarius S, Wildemann B. Aquaporin-4 antibodies (NMO-IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain pathology. 2013;23(6):661-83.
-
Waters PJ, McKeon A, Leite MI, Rajasekharan S, Lennon VA, Villalobos A, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology. 2012;78(9):665-71; discussion 9.
-
Sato DK, Nakashima I, Takahashi T, Misu T, Waters P, Kuroda H, et al. Aquaporin-4 antibody- positive cases beyond current diagnostic criteria for NMO spectrum disorders. Neurology. 2013;80(24):2210-6.
-
Pittock SJ, Lennon VA, Bakshi N, Shen L, McKeon A, Quach H, et al. Seroprevalence of aquaporin-4-IgG in a northern California population representative cohort of multiple sclerosis. JAMA neurology. 2014;71(11):1433-6.
-
Klawiter EC, Alvarez E, 3rd, Xu J, Paciorkowski AR, Zhu L, Parks BJ, et al. NMO-IgG detected in CSF in seronegative neuromyelitis optica. Neurology. 2009;72(12):1101-3.
-
Long Y, Qiu W, Lu Z, Peng F, Hu X. Clinical features of Chinese patients with multiple sclerosis with aquaporin-4 antibodies in cerebrospinal fluid but not serum. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia. 2013;20(2):233-7.
-
Jarius S, Aboul-Enein F, Waters P, Kuenz B, Hauser A, Berger T, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain : a journal of neurology. 2008;131(Pt 11):3072-80.
-
Jarius S, Franciotta D, Paul F, Ruprecht K, Bergamaschi R, Rommer PS, et al. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. Journal of neuroinflammation. 2010;7:52.
-
Jarius S, Franciotta D, Paul F, Bergamaschi R, Rommer PS, Ruprecht K, et al. Testing for antibodies to human aquaporin-4 by ELISA: sensitivity, specificity, and direct comparison with immunohistochemistry. Journal of the neurological sciences. 2012;320(1-2):32-7.
-
Kim W, Lee JE, Li XF, Kim SH, Han BG, Lee BI, et al. Quantitative measurement of anti- aquaporin-4 antibodies by enzyme-linked immunosorbent assay using purified recombinant human aquaporin-4. Multiple sclerosis. 2012;18(5):578-86.
-
Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M, et al. Anti-aquaporin- 4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain : a journal of neurology. 2007;130(Pt 5):1235-43.
Appendix
NMOSD diagnostic criteria for adult patients
Diagnostic criteria for NMOSD with NMO-IgG
-
At least 1 core clinical characteristic
-
Positive test for NMO-IgG using best available detection method (cell-based assay strongly recommended)
-
Exclusion of alternative diagnoses
Diagnostic criteria for NMOSD without NMO-IgG or NMOSD with unknown NMO-IgG status
-
At least 2 core clinical characteristics occurring as a result of one or more clinical attacks and meeting all of the following requirements:
- At least 1 core clinical characteristic must be optic neuritis , acute myelitis with LETM, or area postrema syndrome
- Dissemination in space (2 or more different core clinical characteristics)
- Fulfillment of additional MRI requirements, as applicable
-
Negative tests for NMO-IgG using best available detection method, or testing unavailable
-
Exclusion of alternative diagnoses
Core clinical characteristics
-
Optic neuritis
-
Acute myelitis
-
Area postrema syndrome: episode of otherwise unexplained hiccups or nausea and vomiting
-
Acute brainstem syndrome
-
Symptomatic narcolepsy or acute diencephalic clinical syndrome with NMOSD-typical diencephalic MRI lesions
-
Symptomatic cerebral syndrome with NMOSD-typical brain lesions
Additional MRI requirements for NMOSD without NMO-IgG and NMOSD with unknown NMO-Ig status
-
Acute optic neuritis: require brain MRI showing (a) normal findings or only nonspecific white matter lesions, OR (b) optic nerve MRI with T2-hyperintense lesion or T1-weighted gadolinium-enhancing lesion extending over >1/2 optic nerve length or involving optic chiasm
-
Acute myelitis: requires associated intramedullary MRI lesion extending over >=3 contiguous segments (LETM) OR >=3 contiguous segments of focal spinal cord atrophy in patients with history compatible with acute myelitis
-
Area postrema syndrome: requires associated dorsal medulla/ area postrema lesions 4. Acute brainstem syndrome: requires associated periependymal brainstem lesions
Neurology 2015;85:177-189
Clinical features and laboratory findings atypical for NMOSD, that need to consider alternative diagnoses
-
Progressive overall clinical course (neurologic deterioration unrelated to attacks: Consider MS)
-
Atypical time to attack nadir: less than 4 hours (consider cord ischemia/ infarction); continual worsening for more than 4 weeks from attack onset (consider sarcoidosis or neoplasm)
-
Partial transverse myelitis, especially when not associated with LETM MRI lesion (consider MS)
-
Presence of CSF oligoclonal bands (oligoclonal bands occur in < 20% of cases of NMO vs > 80% of MS
Comorbidities associated with neurologic syndromes that mimic NMOSD
-
Sarcoidosis, established or suggestive clinical, radiologic or laboratory findings thereof (e.g. mediastinal adenopathy, fever and night sweats, elevated serum angiotensin converting enzyme or interleukin-2 receptor level)
-
Cancer, established or with suggestive clinical, radiologic or laboratory findings thereof; consider lymphoma or paraneoplastic disease ( e.g. collapsing response mediator protein-5 associated optic neuropathy and myelopathy or anti-Ma- associated diencephalic syndrome)
-
Chronic infection, established or with suggestive clinical radiologic, or laboratory findings thereof ( e.g. HIV, syphilis)